Rett Syndrome
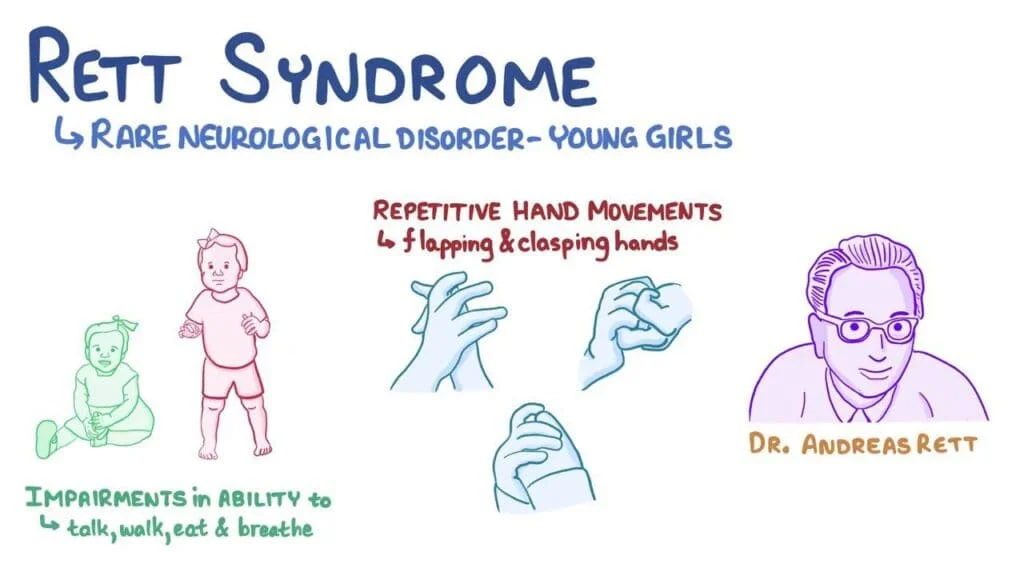
Rett syndrome is a rare genetic neurological disorder that predominantly affects females, with an estimated incidence of 1 in 10,000 to 15,000 live births. First described by Austrian physician Dr. Andreas Rett in 1966, this condition is characterized by normal early development followed by a period of regression, leading to a range of severe cognitive, motor, and communication impairments.
Table of Contents
Definition/Description
Rett syndrome is a rare progressive disorder of the nervous system, leading to impaired cognitive and physical development. The disorder results from a non-inherited genetic mutation, with almost all cases having no family history.
Epidemiology
Rett syndrome occurs almost exclusively in girls, affecting approximately 1 in every 10,000-15,000 females.
The incidence rate in males is unknown, partly due to males with the genetic mutation rarely surviving childbirth.
In the rare circumstance where males with Rett syndrome survive, deficits are often more severe, as males do not have an additional X chromosome to compensate for the mutation.
Children with Rett syndrome typically show normal development until 6 to 18 months after birth, later followed by regression of cognitive, language, and motor function.
On average, the life expectancy of females with Rett syndrome ranges between 40 to 50 years old, with death often occurring unexpectedly or due to secondary causes such as pneumonia.
Causes of Rett Syndrome
Most cases of Rett syndrome are caused by a non-inherited mutation on the dominant X chromosome, on the gene encoding methyl-CpG-binding protein-2 (MECP2).
MECP2 is important for DNA methylation and mutations of this protein result in the inability to deactivate or repress specific genes.
The ability to ‘turn-off’ certain genes is necessary for normal development and maintenance of the nervous system, with impairment in this function potentially leading to cognitive and motor deficits.
The severity of Rett syndrome varies dramatically between individuals and depends on the type and location of the MECP2 mutation, as well as the process of random X chromosome inactivation.
Approximately 5-10% of cases do not appear to have MECP2 mutation. Infants often experiencing seizures before 6 months of age were often diagnosed as having Rett syndrome and were described as atypical cases resulting from mutations in the cyclin-dependent kinase-like 5 (CDKL5) gene, however, CDKL5 has now been classified as a separate disorder.
Further research needs to be conducted in order to investigate how MECP2 gene mutations and other factors contribute to the development and severity of Rett syndrome.
A rare genetic mutation affecting brain development in girls.
Rett syndrome is a rare genetic neurological and developmental disorder that affects the way the brain develops, causing a progressive loss of motor skills and speech. This disorder primarily affects girls.
Most babies with Rett syndrome seem to develop normally for the first 6 to 18 months of age, and then lose skills they previously had such as the ability to crawl, walk, communicate, or use their hands.
Over time, children with Rett syndrome have increasing problems with the use of muscles that control movement, coordination, and communication.
Rett syndrome can also cause seizures and intellectual disability. Abnormal hand movements, such as repetitive rubbing or clapping, replacing objects.
Although there’s no cure for Rett syndrome, potential treatments are being studied.
Current treatment focuses on improving movement and communication, treating seizures, and providing care and support for children and adults with Rett syndrome and their families.
Despite being caused by a gene mutation, Rett syndrome is rarely inherited.
Infants seem healthy during their first six months, but over time, rapidly lose coordination, speech, and use of their hands. Symptoms may then stabilize for years.
There’s no cure, but medication, physio- and speech therapy, and nutritional support help manage symptoms, prevent complications, and improve quality of life.
Symptoms of Rett Syndrome
Rett syndrome is characterized by normal development during the first few months of life followed by regression of motor and communication skills, cognitive impairment, stereotypic hand movements, abnormal breathing, and gait abnormalities.
In Rett syndrome, the central nervous system is primarily affected; however, it often manifests as a multi-system disorder that impacts a child’s growth, pubertal development, and overall health.
Co-morbidities are common in Rett syndrome, including gastrointestinal problems, scoliosis, epilepsy, unusual breathing patterns, sleep disturbances, and low bone density leading to an increased risk of fractures.
Scoliosis is the most prevalent orthopedic co-morbidity, occurring by age 15 in approximately 75% of individuals with Rett syndrome.
Altered sensitivity to pain is another characteristic that individuals with Rett syndrome may experience.
Signs and symptoms of Rett syndrome include
- Developmental and language skills
- Early developmental skills are usually acquired but many later than normal
- Significantly impaired communication and cognitive abilities
- Many children lose the ability to speak at around 12 to 18 months
- Gross motor and receptive language acquisition is often superior to fine motor and expressive language skills
- Mathematics and reading skills are delayed or absent
- Apraxia
- Inability or impaired ability to perform tasks or movements
- Deficits in movements such as eye gaze and speech
- Complex motor skills such as managing stairs or riding a bike are delayed or absent
- Hand movements
- Hand wringing/squeezing, clapping/tapping, mouthing, and washing/rubbing automatisms are stereotypical in Rett syndrome
- Repeatedly moving the hands toward the mouth
- Breathing Irregularities
- Apnea (breath holding)
- Hyperventilation
- Air swallowing
- Other neurological symptoms
- Epilepsy
- Sleep disturbances
- Tremors
- Excess salivation
- Cognitive impairments
- Symptoms affecting other parts of the body
- Scoliosis
- Microcephaly (small head size)
- Small hands and feet
- Gastrointestinal problems, including reflux and constipation
- Teeth grinding, issues with chewing and swallowing
- Heart rhythm abnormalities
- Low muscle tone
- Dystonia
- Toe walking
0THER SYMPTOMS
- Slowed growth. Brain growth slows after birth. Smaller than normal head size (microcephaly) is usually the first sign that a child has Rett syndrome. As children get older, delayed growth in other parts of the body becomes evident.
- Loss of normal movement and coordination.
- The first signs often include reduced hand control and a decreasing ability to crawl or walk normally. At first, this loss of abilities occurs rapidly, and then it continues more gradually. Eventually, muscles become weak or may become rigid or spastic with abnormal movement and positioning.
- Loss of communication abilities. Children with Rett syndrome typically begin to lose the ability to speak, make eye contact, and communicate in other ways.
- They may become disinterested in other people, toys, and their surroundings. Some children have rapid changes, such as a sudden loss of speech.
- Over time, children may gradually regain eye contact and develop nonverbal communication skills.
- Abnormal hand movements.
- Children with Rett syndrome typically develop repetitive, purposeless hand movements that may differ for each person. Hand movements may include hand-wringing, squeezing, clapping, tapping, or rubbing.
- Unusual eye movements.
- Children with Rett syndrome tend to have unusual eye movements, such as intense staring, blinking, crossed eyes, or closing one eye at a time.
- Breathing problems. These include breath-holding, abnormally rapid breathing (hyperventilation), forceful exhalation of air or saliva, and swallowing air. These problems tend to occur during waking hours, but breathing disturbances such as shallow breathing or periodic breathing can occur during sleep.
- Irritability and crying. Children with Rett syndrome may become increasingly agitated and irritable as they get older. Periods of crying or screaming may begin suddenly, for no apparent reason, and last for hours.
- Some children may experience fears and anxiety.
- Other abnormal behaviors. These may include, for example, sudden, odd facial expressions and long bouts of laughter, hand licking, and grasping of hair or clothing.
- Cognitive disabilities. Loss of skills can be accompanied by a loss of intellectual functioning.
- Seizures.
- Most people who have Rett syndrome experience seizures at some time during their lives. Multiple seizure types may occur and are accompanied by an abnormal electroencephalogram (EEG).
- Abnormal curvature of the spine (scoliosis). Scoliosis is common with Rett syndrome. It typically begins between 8 and 11 years of age and increases with age.
- Surgery may be required if the curvature is severe. irregular heartbeat.
- This is a life-threatening problem for many children and adults with Rett syndrome and can result in sudden death.
- Sleep disturbances.
- Abnormal sleep patterns can include irregular sleep times, falling asleep during the day and being awake at night, or waking in the night with crying or screaming.
- Other symptoms.
- A variety of other symptoms can occur, such as thin, fragile bones prone to fractures; small hands and feet that are usually cold; problems with chewing and swallowing;
Stages of Rett syndrome
Rett syndrome is commonly divided into four stages:
Stage I: early onset. Signs and symptoms are subtle and easily overlooked during the first stage, which starts between 6 and 18 months of age and can last for a few months or a year.
Babies in this stage may show less eye contact and start to lose interest in toys. They may also have delays in sitting or crawling.
Stage II: rapid deterioration. Starting between 1 and 4 years of age, children lose the ability to perform skills they previously had.
This loss can be rapid or more gradual, occurring over weeks or months. Symptoms of Rett syndrome occur, such as slowed head growth, abnormal hand movements, hyperventilating, screaming or crying for no apparent reason, problems with movement and coordination, and a loss of social interaction and communication.
Stage III: plateau. The third stage usually begins between the ages of 2 and 10 years and can last for many years.
Although problems with movement continue, behavior may have limited improvement, with less crying and irritability, and some improvement in hand use and communication.
Seizures may begin in this stage and generally don’t occur before the age of 2 Year
Stage IV: late motor deterioration. This stage usually begins after the age of 10 and can last for years or decades.
It’s marked by reduced mobility, muscle weakness, joint contractures, and scoliosis. Understanding, communication, and hand skills generally remain stable or improve slightly, and seizures may occur less often.