
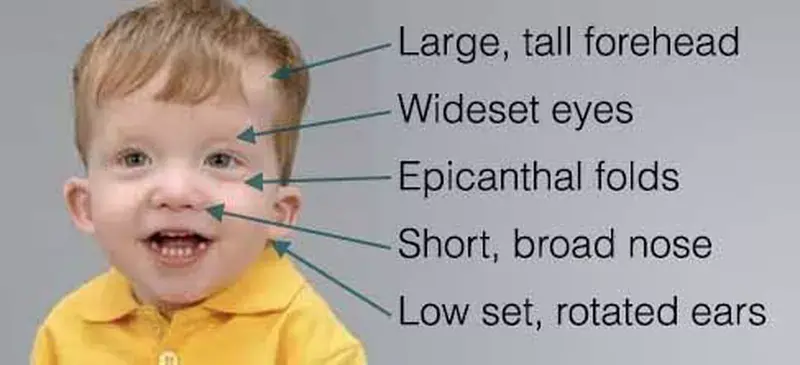
Noonam Syndrome
Table of Contents
What is Noonan Syndrome?
A hereditary condition known as Noonan syndrome stops some body parts from developing normally. There are many different ways that Noonan syndrome can impact an individual.
These include small stature, heart disorders, various physical issues, odd face features, and potential developmental delays.
Noonan syndrome is caused by a genetic mutation and is inherited when a kid inherits a copy of the defective gene from a parent (dominant inheritance).
Additionally, it may arise as a spontaneous mutation, in which case no family history is relevant.
Controlling the symptoms and complications of Noonan syndrome is the main goal of management. Some individuals with Noonan syndrome may benefit from growth hormone treatment for their small stature.
Symptoms
Signs and symptoms of Noonan syndrome vary greatly among individuals and may be mild to severe. Characteristics may be related to the specific gene containing the mutation.
Facial features
Facial appearance is one of the key clinical features that leads to a diagnosis of Noonan syndrome.
These features may be more pronounced in infants and young children, but change with age. In adulthood, these distinct features become more subtle.
Features may include the following:
- Eyes are wide-set and down-slanting with droopy lids. Irises are pale blue or green.
- Ears are low-set and rotated backward.
- The nose is depressed at the top, with a wide base and bulbous tip.
- The mouth has a deep groove between the nose and the mouth and wide peaks in the upper lip. The crease that runs from the edge of the nose to the corner of the mouth becomes deeply grooved with age.
- Teeth may be crooked, the inside roof of the mouth (palate) may be highly arched and the lower jaw may be small.
- Facial features may appear coarse, but appear sharper with age. The face may appear droopy and expressionless.
- The Head may appear large with a prominent forehead and a low hairline on the back of the head.
- The skin may appear thin and transparent with age.
- Heart disease
- Many people with Noonan syndrome are born with some form of heart defect (congenital heart disease), accounting for some of the key signs and symptoms of the disorder.
- Some heart problems can occur later in life. Some forms of congenital heart disease associated with this disorder include,
Valve disorders. Pulmonary valve stenosis is a narrowing of the pulmonary valve, the flap of tissue that separates the lower right chamber (ventricle) of the heart from the artery that supplies blood to the lungs (pulmonary artery).
It’s the most common heart problem seen with Noonan syndrome, and it may occur alone or with other heart defects.
Thickening of the heart muscle (hypertrophic cardiomyopathy). This is abnormal growth or thickening of the heart muscle that affects some people with Noonan syndrome.
Other structural defects of the heart. The defects can involve a hole in the wall that separates the two lower chambers of the heart (ventricular septal defect), narrowing of the artery that carries blood to the lungs for oxygen (pulmonary artery stenosis), or narrowing of the major blood vessel (aorta) that carries blood from the heart to the body (aortic coarctation).
Irregular heart rhythm.
This can occur with or without structural heart abnormalities. Irregular heart rhythm occurs in the majority of people with Noonan syndrome.
Growth issues.
Noonan syndrome can affect normal growth. Many children with Noonan syndrome don’t grow at a normal rate. Issues may include the following:
- Birth weight will likely be normal, but growth slows over time.
- Eating difficulties may result in inadequate nutrition and poor weight gain.
- Growth hormone levels may be insufficient.
- The growth spurt that’s usually seen during the teenage years may be delayed. But because this disorder causes bone maturity to be delayed, growth sometimes continues into the late teens.
- By adulthood, some people with Noonan syndrome may have normal height, but short stature is more common.
- Musculoskeletal issues
- Some common issues can include:
- An unusually shaped chest often with a sunken sternum (pectus excavatum) or raised sternum (pectus carinatum)
- Wide-set nipples
- Short neck, often with extra folds of skin (webbed neck) or prominent neck muscles (trapezius muscle)
- Deformities of the spine
- Learning disabilities
- Intelligence isn’t affected for most people with Noonan syndrome. However, individuals may have:
- An increased risk of learning disabilities and mild intellectual disability
- A wide range of mental, emotional, and behavioral issues that are usually mild
- Hearing and vision deficits that may complicate learning
- Eye conditions
- A common sign of Noonan syndrome is abnormalities of the eyes and eyelids. These may include:
- Problems with the eye muscles, such as cross-eye (strabismus)
- Refractive problems, such as astigmatism, nearsightedness (myopia), or farsightedness (hyperopia)
- Rapid movement of the eyeballs (nystagmus)
- Cataracts
- Hearing problems
- Noonan syndrome can cause hearing deficits due to nerve issues or structural abnormalities in the inner ear bones.
Bleeding
Noonan syndrome can cause excessive bleeding and bruising due to clotting defects or having too few platelets.
Lymphatic conditions
Noonan syndrome can cause problems with the lymphatic system, which drains excess fluid from the body and helps fight infection. These problems:
- May show up before or after birth or develop in the teenage years or adulthood
- Can be focused on a particular area of the body or widespread
- Most commonly cause excess fluid (lymphedema) on the back of the hands or top of the feet
- Genital and kidney conditions
- Many people, especially males, with Noonan syndrome can have problems with the genitals and kidneys.
Testicles. Undescended testicles (cryptorchidism) are common in males.
Puberty. ;
Puberty may be delayed in both boys and girls.
Fertility. Most females develop normal fertility. In males, however, fertility may not develop normally, often because of undescended testicles.
Kidneys. ;
Kidney probzlems are generally mild and occur in a fairly small number of people with Noonan syndrome.
Skin conditions
People with Noonan syndrome may have skin conditions,
which most commonly are: Various problems that affect the color and texture of the skinCurly, coarse hair, or sparse hair
Causes
Noonan syndrome may result from a mutation in one or more genes. Proteins produced by mutations in these genes are constantly active. This ongoing protein activation interferes with the normal process of cell development and division because these genes are involved in the formation of tissues in the body.
Noonan syndrome may result from the following gene alterations:
Inherited.
Children who have a parent with Noonan syndrome who carries the altered gene are 50% more likely to get the disorder themselves. This form of inheritance is known as autosomal dominant.
Random.
A child may get Noonan syndrome if they have a newly altered gene, which means they did not receive it from a parent. We call this a de novo genetic disease.
The cause of Noonan syndrome is unknown in many circumstances.
Pathophysiology
Because Noonan syndrome involves a mutation in the RAS/MAPK pathway, it is categorised as a RASopathy.
Ras/mitogen-activated protein kinase (MAPK) pathway germline mutations are the source of hereditary diseases known as RASopathies.
Neurofibromatosis type 1 (NF1), cardiofaciocutaneous syndrome, LEOPARD syndrome, and Costello syndrome are some of these disorders.
Migration, differentiation, proliferation, and cell division all depend on the Ras/MAPK pathway. It is essential for normal growth.
Cell division, proliferation, differentiation, and migration all depend on the Ras/MAPK pathway. It is essential for normal development. Most often, a mutation in the PTPN11 gene results in Noonan syndrome. A lesser percentage of mutations are seen in RIT1, SOS1, and RAF1. There are three types of PTPN11 mutations: de novo, autosomal dominant, and sporadic. The mutations that cause Noonan syndrome are regarded as gain of function mutations because they cause the RAS/MAPK signalling to be inappropriately prolonged. In Noonan syndrome, the pleomorphic traits are caused by the extension of the RAS/MAPK pathway.
Risk Factor
A kid of a parent with Noonan syndrome has a 50% chance of inheriting the mutated gene. It’s possible that the child who receives the faulty gene will experience greater symptoms than the parent.
Complications
There may be Noonan syndrome complications that require care, such as:
Delays in development. It’s possible that children with Noonan syndrome develop more slowly than other kids their age. For instance, kids could lag far behind in school-related learning or in learning to walk or talk. The children’s learning and educational demands, as well as their developmental challenges, require a strategy.
Bruise and bleeding. The common bleeding problem in persons with Noonan syndrome is sometimes not identified until dental procedures or surgery.
Buildup of fluid.
This issue, known as lymphedema, is characterized by an excess of fluid that accumulates in different parts of the body. The area surrounding the heart and lungs may occasionally fill with fluid.
Urinary tract issues.
Urinary tract infections may become more likely if the kidney structure is abnormal.
Fertility issues.
Poor sperm counts and other fertility problems may be experienced by males with undescended testicles or testicles that are dysfunctional.
Higher risk of cancer.
For example, leukemia or certain tumors may be more likely to develop than other cancers.
Prevention
Discuss the advantages of genetic counseling with your physician or healthcare team prior to having children if you have a family history of Noonan syndrome. There is a genetic test for Noonan syndrome.
Early detection of Noonan syndrome may reduce consequences like heart disease if appropriate and continuous care is provided.
Diagnosis
Noonan syndrome is usually diagnosed by a physician after observing some important symptoms. However, this can be challenging because some of the condition’s characteristics are difficult to identify and locate.
Noonan syndrome can occasionally go undetected until maturity, following the birth of a child who is more obviously impacted by the disorder. A diagnosis can be verified by genetic testing.
The kind and severity of heart abnormalities can be determined by a cardiologist if there is evidence of them.
Treatment
Noonan syndrome has no known cure, however there are therapies that can lessen its consequences. The benefits increase with the timing of diagnosis and therapy.
The severity of the symptoms and effects determines how Noonan syndrome is treated. The same care is given to many of the physical and health problems as to everyone else. A coordinated team approach is the most effective way to address the numerous issues associated with this medical condition.
Some such methods could be:
Heart treatment.
Some types of cardiac issues may be treated with specific medications. Surgery can be required if there is an issue with the heart’s valves. Occasionally, the doctor may also recommend that heart function be assessed.
Treating low growth rate.
Up to age three, a health care provider should take a patient’s height three times a year; after that, they should do it once a year until they are an adult. Your child will grow as a result of this. Blood tests could be prescribed to determine whether there is a nutritional issue. Growth hormone therapy can be a possibility if your child’s growth hormone levels aren’t high enough.
Handling difficulties with learning.
Consult your physician or other healthcare provider about infant stimulation programs if you have developmental delays in your early years. Speech and physical therapy may be required. Special education or teaching methods adapted to your child’s requirements may be suitable in some situations.
Vision and hearing treatments.
It is advised to have eye exams at least every two years. Most eye problems can be resolved with just glasses. For certain disorders, such cataracts, surgery may be required. Every year in childhood, hearing tests are advised.
Treatment for bleeding and bruising.
If there is a history of bleeding problems or easily becoming bruised, stay away from aspirin and items that contain it. Doctors may occasionally recommend medications that aid in blood clotting. Before undergoing any operations, inform medical personnel of any bleeding or bruises.
Treatment for fluid buildup.
Treatment for fluid accumulation in the body may or may not be necessary. Discuss this with your physician or medical team. They might advise taking particular actions.
Treatment for genital problems.
Your child may require surgery if, during the first several months of life, one or both testicles have not shifted into the correct position.
Additional assessments and ongoing follow-up care might be required. Certain issues determine this. It is recommended that individuals with Noonan syndrome have ongoing medical monitoring on a regular basis.
Support
Consult your healthcare staff about available local support groups. Asking about reliable internet sites that can connect you to nearby support groups and information about Noonan syndrome is another option.
Getting ready for your scheduled appointment
Consult your child’s paediatrician or primary care physician if you believe you or your child may have Noonan syndrome. However, a specialist can be suggested based on the symptoms, such as a physician with expertise in genetics or heart issues.
To help you prepare for your appointment, here are some details. If at all possible, bring a family member or friend. Memory retention and emotional support might be provided by a trustworthy companion.
Epidemiology
Noonan syndrome affects roughly 1 in 1000 to 2500 people. Both boys and females are equally impacted because of the autosomal dominant mode of inheritance. Cryptorchidism may manifest in males, allowing for an earlier diagnosis. Noonan syndrome is equally prevalent in all ethnic groups.
Differential Diagnosis
There are many potential possible diagnoses for Noonan syndrome. A comparable set of symptoms, including congenital heart disease, small height, and developmental delay, can be caused by numerous additional chromosomal disorders.
Turner syndrome
Considering the similarities between Noonan and Turner syndromes, Noonan syndrome has been called “pseudo-Turner syndrome.”
Both disorders are characterised by short height and a webbed neck. But Turner syndrome (45, X0) only affects women since it affects the X chromosome. The karyotype of Noonan syndrome is normal, and it affects both boys and girls.
Since the RAS/MAPK system is impacted by the genetic abnormalities, Noonan syndrome is regarded as a RASopathy. Other RASopathies share traits with Noonan syndrome.
Among these conditions are:
- Cardiofasciocutaneous syndrome
- Costello syndrome
- Neurofibromatosis 1
- Noonan syndrome with multiple lentigines (LEOPARD syndrome)
Prognosis
The severity of the phenotypic in people with Noonan Syndrome determines their prognosis. A correlation exists between the severity of the heart defect and patient mortality and morbidity. Many patients have little morbidity and an average lifetime.
Team of Doctors
An interprofessional team of a geneticist, paediatrician, primary care physician, ENT surgeon, audiologist, ophthalmologist, and cardiologist is necessary for the diagnosis and treatment of Noonan syndrome. Since Noonan syndrome is a genetically inherited condition, there is no known treatment. The management of Noonan syndrome is to relieve symptoms and provide supportive care. Multiple organ systems must be treated, and interprofessional treatment is frequently required.
Throughout childhood, ophthalmology and hearing testing are appropriate. If the testes have not descended in men with cryptorchidism, orchiopexy should be done when the infant is around one year old. The purpose of this surgery is to lower the adult risk of testicular cancer. An echocardiogram and electrocardiogram should be acquired for congenital cardiac defects. Every five years, a cardiac assessment is required, even for those who have not been diagnosed with heart abnormalities. For growth hormone therapy, short stature might be assessed. Targeting lymphoedema can be accomplished with the right supporting measures.
FAQs
Short stature, congenital cardiac abnormalities, and distinctive facial dysmorphology are the three characteristics that define Noonan syndrome. Features such as widely separated eyes, low-set ears, and a webbed neck are examples of facial dysmorphology. Although these are the main characteristics, other physical and developmental problems can also affect people with Noonan syndrome.
While the majority of people with Noonan syndrome are intelligent, some may have intellectual disabilities or specific educational needs. Both immunological response and the body’s ability to get essential nutrients depend heavily on the lymphatic system.
Eight male patients with Noonan’s syndrome, ages 13 to 26, had median IQs of 102, with a range of 64 to 127. The potential existence of a particular verbal or praxic (visual-constructional) handicap was concealed by the full IQ.
Patients with Williams-Beuren syndrome have a broader mouth with a thicker lower lip, a shorter nose with longer alas, and substantially more down-slanted palpebral fissures than patients with Noonan syndrome, who have far more marked hypertelorism and telecanthus.
References
- Noonan syndrome – Symptoms and causes. (n.d.). Mayo Clinic. https://www.mayoclinic.org/diseases-conditions/noonan-syndrome/symptoms-causes/syc-20354422
- Allen, M. J., & Sharma, S. (2023, January 9). Noonan Syndrome. StatPearls – NCBI Bookshelf. https://www.ncbi.nlm.nih.gov/books/NBK532269/
2 Comments